Maladie de Hirschsprung

Chaque année, des parents aux quatre coins du monde accueillent avec joie un nouveau membre dans leur famille. Mais parfois, au milieu de ces moments heureux, des inquiétudes surgissent. Certains bébés, pour des raisons mystérieuses, semblent avoir du mal à manger, à prendre du poids ou même à avoir des selles normales. Parmi ces cas, une maladie rare mais sérieuse peut être en cause : la maladie de Hirschsprung. Mais qu’est-ce que c’est exactement ? Et comment ce trouble, qui affecte le système digestif, peut-il changer la vie d’un enfant et de sa famille ? Plongeons ensemble dans l’univers de cette maladie méconnue pour en comprendre les enjeux et les traitements.
 Qu’est-ce que ?
Qu’est-ce que ?
Définition
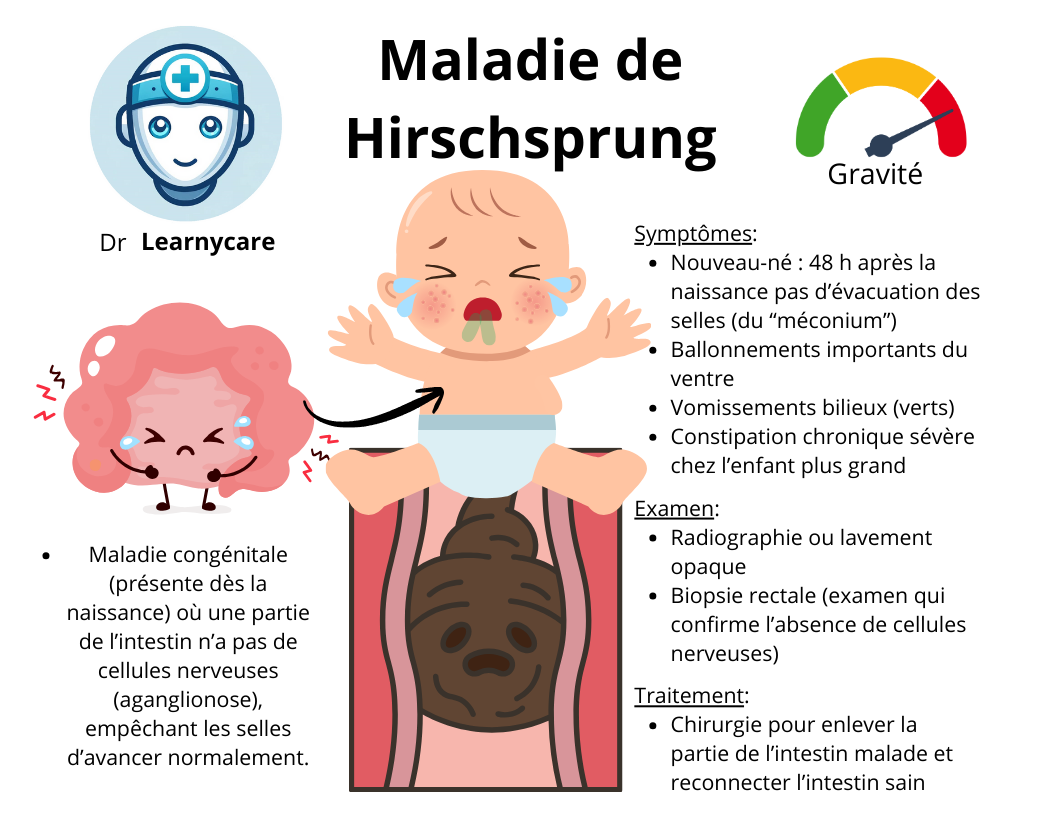
La maladie de Hirschsprung, également connue sous le nom de mégacôlon congénital, est une affection présente dès la naissance. Elle est caractérisée par l’absence de cellules nerveuses spécifiques, appelées cellules ganglionnaires, dans certaines parties de l’intestin. Voici une définition approfondie :
La maladie de Hirschsprung est une anomalie congénitale du système nerveux entérique, qui se manifeste par l’absence de cellules ganglionnaires dans le système digestif. Ces cellules sont essentielles car elles régulent les mouvements intestinaux qui permettent la progression des selles. Lorsqu’elles sont absentes, cela entraîne une zone d’intestin non fonctionnelle qui ne se contracte pas normalement, ce qui peut provoquer une obstruction.
La partie la plus couramment affectée est le rectum et le bas sigmoïde, bien que la maladie puisse parfois s’étendre à d’autres parties du côlon. Dans environ 5% des cas, l’ensemble du colon est touché. Plus rarement, soit dans environ 1% des cas, la maladie peut également affecter l’intestin grêle.
Explications
La physiopathologie de la maladie de Hirschsprung est complexe et a été l’objet de nombreuses études. Comprendre comment cette maladie se développe est essentiel pour appréhender ses symptômes et son traitement. Explorons ensemble ces mécanismes sous-jacents.
Échec de la migration des neuroblastes :
La formation du système nerveux intestinal est un processus complexe qui implique la migration des cellule neurologiques durant le développement embryonnaire.
Dans la maladie de Hirschsprung, cette migration échoue, empêchant la colonisation complète du côlon par les cellules ganglionnaires. Cette absence est typiquement observée dans le rectum et peut s’étendre de manière proximale vers le colon et, dans de rares cas, l’intestin grêle.
Anomalies génétiques :
Plusieurs gènes ont été associés à la maladie de Hirschsprung. Des mutations dans ces gènes peuvent perturber le développement ou la fonction du système nerveux entérique.
Parmi ces gènes, les plus couramment impliqués sont RET, EDNRB, et SOX10, mais d’autres peuvent aussi jouer un rôle. La présence de mutations génétiques peut influencer l’étendue de la maladie et être associée à d’autres anomalies congénitales.
Hypopéristaltisme du segment atteint :
Les cellules ganglionnaires jouent un rôle crucial dans la coordination des contractions péristaltiques qui propulsent les matières fécales à travers le côlon. En leur absence, la partie affectée de l’intestin devient akinétique, c’est-à-dire qu’elle ne se contracte pas normalement.
Cette absence de mouvements coordonnés conduit à un hypopéristaltisme ou à une absence totale de péristaltisme dans le segment atteint.
Dilatation du côlon en amont :
L’obstruction fonctionnelle causée par le segment akinétique empêche les matières fécales de progresser normalement, provoquant leur accumulation en amont.
Cette accumulation entraîne une dilatation du côlon en amont du segment affecté, conduisant à ce que l’on appelle un mégacôlon.
Ces mécanismes physiopathologiques expliquent la sévérité des symptômes observés chez les patients atteints de la maladie de Hirschsprung. La reconnaissance et la prise en charge précoces sont cruciales pour prévenir les complications associées à cette affection.
 Diagnostic
Diagnostic
Les personnes concernées
L’épidémiologie se concentre sur l’étude de la fréquence, de la distribution et des déterminants des maladies dans les populations. Pour la maladie de Hirschsprung, voici une synthèse épidémiologique :
Population affectée :
La maladie est généralement diagnostiquée chez le jeune enfant ou le nouveau-né. Bien qu’elle soit présente dès la naissance, les symptômes peuvent ne pas être immédiatement apparents.
Prédominance masculine :
Les garçons sont disproportionnellement affectés, représentant environ 80 % des cas. Cela suggère une composante génétique ou chromosomique qui pourrait influencer la susceptibilité à cette maladie.
Symptômes initiaux :
Un indicateur courant chez les nouveau-nés est le retard à l’évacuation du méconium, qui est la première selle d’un bébé. Si un nouveau-né ne passe pas de méconium dans les 24 heures suivant la naissance, cela peut être un signe de la maladie de Hirschsprung.
Incidence :
La maladie de Hirschsprung est relativement rare, affectant environ 1 sur 5 000 naissances vivantes. Cela signifie qu’elle touche environ 0,02 % des nouveau-nés.
Facteurs de risque :
Antécédents familiaux : la maladie de Hirschsprung peut avoir une composante héréditaire. Les enfants ayant un frère, une sœur, un parent ou un autre membre de la famille atteint de la maladie ont un risque accru de développer cette affection. Cependant, la plupart des cas sont sporadiques, c’est-à-dire qu’ils se produisent sans antécédents familiaux connus.
La connaissance de ces données épidémiologiques peut aider les professionnels de santé à mieux reconnaître et diagnostiquer cette maladie, et à informer les familles des risques potentiels si elles ont des antécédents familiaux.
Les symptômes
La maladie de Hirschsprung, en affectant le système nerveux entérique, entraîne une série de symptômes gastro-intestinaux. Ces symptômes peuvent varier en fonction de l’âge de l’individu et de l’étendue de l’intestin qui est touché. Voici une description des symptômes les plus courants :
- Constipation rebelle :
- La constipation est le symptôme le plus courant et le plus persistant de la maladie de Hirschsprung. Elle est dite “rebelle” car elle ne répond pas bien aux traitements classiques de la constipation. Les selles peuvent être absentes pendant plusieurs jours, voire plus longtemps. Cette constipation est due à l’absence de mouvements péristaltiques efficaces dans le segment d’intestin touché par la maladie.
- Ballonnement :
- En raison de l’obstruction fonctionnelle, les gaz et les selles peuvent s’accumuler, provoquant un ballonnement abdominal. Ce ballonnement peut être douloureux et entraîner un inconfort important pour l’enfant.
- Vomissements :
-
- Dans les cas plus sévères, ou si l’obstruction est plus étendue, les enfants peuvent présenter des vomissements. Ces vomissements peuvent contenir de la bile (de couleur verte) et se produisent généralement lorsque les matières fécales ne peuvent pas progresser normalement à travers le système digestif.
D’autres symptômes peuvent également apparaître chez certains enfants, comme une perte de poids, une faiblesse ou un retard de croissance, surtout si la maladie n’est pas diagnostiquée et traitée rapidement. Il est essentiel pour les parents et les professionnels de santé de reconnaître ces signes et d’agir en conséquence pour éviter des complications potentiellement graves.
Les signes
Les signes cliniques se réfèrent aux manifestations observables et mesurables de la maladie, souvent identifiées par un examen physique par un professionnel de la santé. Dans le cas de la maladie de Hirschsprung, voici les signes cliniques typiques :
- Généraux :
- Retard staturo-pondéral : les enfants atteints peuvent présenter un retard de croissance, ne gagnant pas de poids ni de taille au même rythme que leurs pairs. Ce retard peut être dû à une malnutrition secondaire à une mauvaise absorption des nutriments ou à une ingestion réduite en raison de symptômes gastro-intestinaux.
- Digestifs :
- Examen du toucher rectal (TR) :
- Ampoule rectale vide : lors d’un examen du toucher rectal, le médecin peut constater une absence ou une diminution des matières fécales dans l’ampoule rectale, qui devrait normalement contenir des selles. Ceci est indicatif de l’absence de mouvements intestinaux propulsifs dans cette région.
- Débâcle lors du retrait : lorsque le doigt du médecin est retiré après l’examen du toucher rectal, il peut y avoir une expulsion soudaine de gaz et de selles accumulés derrière la zone touchée, phénomène connu sous le nom de “débâcle”. C’est un signe clinique très évocateur de la maladie de Hirschsprung.
- Examen du toucher rectal (TR) :
Il est important de noter que le diagnostic de la maladie de Hirschsprung ne se base pas uniquement sur ces signes cliniques. Des investigations supplémentaires, comme une biopsie rectale, sont nécessaires pour confirmer le diagnostic. Toutefois, ces signes cliniques peuvent orienter le médecin vers la possibilité de cette maladie et justifier des examens plus approfondis.
Les complications
La maladie de Hirschsprung peut se manifester par diverses complications.
- Syndrome occlusif :
- Il s’agit d’une obstruction intestinale aiguë. Les symptômes incluent des douleurs abdominales sévères, des vomissements, une absence de passage de gaz et de selles, et un ballonnement. C’est une urgence médicale qui nécessite une prise en charge rapide.
- Sepsis lié à la colite :
- La stase intestinale peut conduire à une inflammation aiguë du côlon, appelée colite. Si elle n’est pas traitée, elle peut entraîner une infection bactérienne du sang, ou sepsis, qui est une urgence médicale. Les symptômes peuvent inclure de la fièvre, des frissons, une accélération du rythme cardiaque, et une respiration rapide.
Associations avec d’autres maladies génétiques :
La maladie de Hirschsprung peut parfois être associée à d’autres affection génétiques. Environ 25% des patients atteints de cette maladie peuvent également présenter une autre maladie génétique, notamment :
- Syndrome de Down (trisomie 21) : il s’agit de l’affection la plus couramment associée à la maladie de Hirschsprung.
- Autres syndromes : d’autres anomalies chromosomiques ou syndromes génétiques peuvent également être associés, bien que plus rarement.
La prise en charge de la maladie de Hirschsprung nécessite une approche multidisciplinaire pour traiter non seulement les symptômes gastro-intestinaux, mais aussi les éventuelles affections associées. Une évaluation génétique peut être recommandée pour certains patients, en particulier ceux qui présentent d’autres signes ou symptômes évocateurs d’une maladie génétique.
Les examens
La maladie de Hirschsprung est suspectée cliniquement sur la base des symptômes et signes décrits précédemment. Cependant, pour confirmer le diagnostic, il est nécessaire de réaliser une série d’examens. Voici une description détaillée de ces examens :
- Biologie :
- Elle est généralement normale, signifiant qu’il n’y a pas d’anomalies significatives dans les paramètres sanguins couramment analysés.
- Abdomen sans préparation (ASP) :
- Bien qu’il ne permette pas de poser un diagnostic définitif, l’ASP peut montrer certaines anomalies :
- Niveaux hydro-aériques : ils suggèrent une obstruction intestinale.
- Absence d’air au niveau du rectum : c’est une caractéristique de la maladie.
- Pneumatose : il s’agit d’air dans la paroi intestinale, un signe grave qui suggère une nécrose intestinale.
- Bien qu’il ne permette pas de poser un diagnostic définitif, l’ASP peut montrer certaines anomalies :
- Lavement opaque :
- Il permet d’observer le côlon à l’aide d’un produit de contraste :
- Disparité de calibre : la zone malade (généralement recto-sigmoïdienne) est souvent étroite en comparaison avec le segment dilaté du côlon en amont.
- Il permet d’observer le côlon à l’aide d’un produit de contraste :
- Manométrie rectale :
- Cet examen évalue la pression à l’intérieur du rectum :
- Absence de réflexe recto-anal : dans une situation normale, la distension rectale provoque une relaxation du sphincter anal. L’absence de cette relaxation est évocatrice de la maladie de Hirschsprung, bien que cela ne prouve pas le diagnostic.
- Cet examen évalue la pression à l’intérieur du rectum :
- Biopsie rectale :
-
- C’est l’examen définitif pour le diagnostic de la maladie de Hirschsprung :
- Agénésie ganglionnaire : l’absence de cellules ganglionnaires dans la paroi intestinale est la caractéristique de la maladie.
- Épaississement des filets nerveux : lorsqu’ils sont colorés par l’ACE (cholinestérase), ils montrent une hyperplasie schwanienne, signifiant une prolifération des cellules de Schwann.
- C’est l’examen définitif pour le diagnostic de la maladie de Hirschsprung :
La combinaison des symptômes cliniques avec les résultats des examens ci-dessus permet d’orienter et de confirmer le diagnostic de la maladie de Hirschsprung. Une fois le diagnostic confirmé, la prise en charge médicale et chirurgicale peut être planifiée en conséquence.
 Le traitement
Le traitement
Étape 1 : prendre en charge
La prise en charge de la maladie de Hirschsprung nécessite une approche multidisciplinaire. Plusieurs professionnels de santé sont impliqués pour garantir un diagnostic précis, un suivi adéquat et une intervention chirurgicale si nécessaire. Ci-dessous, une vue d’ensemble des différents acteurs impliqués et de leurs rôles respectifs :
- Professionnels de santé de ville :
- Ils jouent souvent un rôle crucial dans les premières étapes de la détection de la maladie. Ce sont généralement les premiers points de contact pour les parents inquiets.
- Ils orientent les patients vers des spécialistes pour des investigations plus approfondies.
- Ils peuvent également conseiller les parents sur les symptômes à surveiller et la gestion initiale.
- Médecin généraliste :
- Il est souvent le premier professionnel de santé à évaluer l’enfant.
- Il est en mesure de poser un diagnostic préliminaire basé sur les symptômes et l’histoire médicale.
- Il oriente ensuite l’enfant vers un spécialiste pour un avis et une confirmation diagnostiques.
- Gastroentérologue :
- Il s’agit d’un spécialiste des maladies du système digestif.
- Il apporte un avis spécialisé sur le diagnostic, la gestion et le traitement.
- Il peut réaliser des investigations supplémentaires, comme une manométrie rectale ou une biopsie, pour confirmer le diagnostic.
- Service de réanimation :
- Dans le cas d’une complication grave comme une entérocolite (inflammation aiguë du côlon), l’enfant pourrait nécessiter une prise en charge en unité de réanimation.
- Ici, l’enfant bénéficiera d’une surveillance étroite et d’une intervention médicale d’urgence pour gérer la septicémie, la déshydratation ou d’autres complications associées.
En plus des professionnels de santé mentionnés ci-dessus, d’autres spécialistes tels que des chirurgiens pédiatriques peuvent être impliqués, notamment pour la chirurgie correctrice. Les psychologues et les conseillers génétiques peuvent également jouer un rôle important pour soutenir les familles face à cette affection. La prise en charge globale vise à offrir à l’enfant la meilleure qualité de vie possible et à éviter les complications potentiellement graves associées à la maladie.
Étape 2 : soulager les symptômes
Le traitement de la maladie de Hirschsprung dépend de la sévérité des symptômes et de l’étendue de la maladie. Avant d’envisager une intervention chirurgicale définitive, il est essentiel de stabiliser l’enfant et de traiter les symptômes aigus. Voici les principales étapes du traitement symptomatique :
1. Réanimation liquidienne
2. Evacuation des selles et des gaz
3. Nutrition
4. Surveillance étroite
Il est important de souligner que le traitement symptomatique est une mesure temporaire pour stabiliser l’enfant et le préparer à une intervention chirurgicale définitive, qui est le seul traitement curatif de la maladie de Hirschsprung. Une fois l’enfant stabilisé, la discussion se portera sur la meilleure approche chirurgicale pour sa situation.
Étape 3 : soigner
Le traitement curatif de la maladie de Hirschsprung vise à éliminer la portion de l’intestin qui ne contient pas de cellules ganglionnaires (portion aganglionnaire) afin de permettre un transit intestinal normal.
 Évolution
Évolution
La maladie de Hirschsprung, une fois diagnostiquée et traitée, présente une évolution généralement favorable. Cependant, le pronostic dépend du diagnostic précoce, du traitement approprié et de la gestion postopératoire des complications potentielles. Voici un aperçu de l’évolution typique de la maladie :
Après un traitement curatif :
La majorité des enfants opérés pour la maladie de Hirschsprung se rétablissent bien et mènent une vie normale sans symptômes persistants.
Le temps nécessaire pour retrouver un fonctionnement intestinal normal varie d’un enfant à l’autre, certains nécessitant des mois pour développer une continence complète.
En résumé, bien que la maladie de Hirschsprung soit une affection grave nécessitant une intervention chirurgicale, la majorité des patients mènent une vie normale après le traitement. Le suivi régulier et une gestion proactive des symptômes ou des complications améliorent le pronostic à long terme.
Aller plus loin
Constipation sévère chez l’enfant, ballonnements… Et si c’était la maladie de Hirschsprung ? Découvrez les signes pour agir tôt et soulager votre enfant.
